Arginine Deprivation Addresses the Achilles Heel of Cancer: Urea Cycle
Deficiencies in Melanoma, HCC and other Cancers
Arginine remains the focus of interest in amino acid deprivation for the treatment of cancer. Intensive investigations both in vitro and in vivo reveal that malignant cells of many types are sensitive to arginine deficiency, the broadening spectrum now including lung, kidney, prostate, mesothelial and breast tumours. Their susceptibility is due to the lack of expression of one or more of the urea cycle enzymes compared with their normal counterparts. Alone or in combination with other chemotherapeutic modalities, arginine deprivation offers a new dimension in the control of tumour growth.
Introduction
Attempts have been made to curtail cancer growth by removing amino acids since the 1950s, but with only limited success and little enthusiasm after a promising start [1]. The pioneers chose to explore phenylalanine, arginine, and methionine [1,2]. The problems were lack of suitable techniques, use of bovine enzyme, and poor understanding of the regulation of the cell cycle in tumour cells compared with normal cells, simply because at that time little was known about it. The only enzyme used regularly in cancer therapy has been L-asparaginase, which set a precedent in its use against leukaemia [3]. A revival of interest has occurred over the last 10-15 years in other enzymes, with a renewed focus on arginine [e.g. 4,5].
In the 1970s, the observation that cells failed to thrive in mycoplasma-infected cultures was attributed to the ability of these organisms to secrete an enzyme, L-arginine deiminase (ADI) into the medium, producing a similar effect to that found with liver extract treatment, attributed to another arginine-catabolising enzyme, L-arginase 1 (Arg1) [discussed in 6]. Inhibition of growth of melanoma, hepatocellular carcinoma and leukaemia were seen with ADI, much as originally found in 1953 by Bach』s group with bovine arginase [7]. These findings were instrumental in the present revival of interest in arginine deprivation. More recently, the number of clinical trials is increasing as new experimental work finds more tumour types that are likely to be good responders. To give two recent examples, the latest findings from Korea have shown the almost complete absence of AS in renal carcinoma cells [8], although clinical efficacy of arginine deprivation has yet to be established. And second, clinical trials are about to begin in London, with main focus on mesothelioma [9].
For other more complex reasons than this, arginine has been adopted as the target amino acid [10], despite its highly complex metabolism and concern that its almost total withdrawal would not be tolerated for any significant period of time by man. Its semi-essential nature should have been seen earlier as a logical target, since arginine amino acid is required at relatively high levels in the plasma during active growth phases when there is a greater demand for it, as also in rapid tumour growth. A better understanding of the physiology, biochemistry, genetics and metabolics of arginine have shown how to keep this particular amino acid under strict control so that we can effectively 「strave」 a tumour to death without the same devastating effect being wrought on normal cells – a perennial problem with conventional chemotherapeutic protocols. [A brief survey of these matters can found in the first issue of Oncology News (www.oncologynews.biz).]
A rational approach in exploiting a frequent difference between normal and tumour cells
The exploitable differential stems from the fact that normal cells move out of cycle into G0 (quiescence) during periods of amino acid starvation, to return to cycle quickly on restoration of the missing amino acid. In contrast, ~80% of tumour cell types have defective restriction point regulation in G1 of the cell cycle [11], continuing to progress – abortively so- under deprivation conditions, often coming up against p53 aberrations that send them down the apoptotic pathway as one form of demise. It is noteworthy that, without any further intervention, arginine deficiency can kill almost all cancer cells within 2-6 days, which gives a reasonable time-frame or therapeutic 「window」 in which to treat sensitive tumours.
The present challenge is to make this deprivation stratefy work to our advantage in cinical oncology. The chosen method has been to produce an arginine deficient state by use of one of the two arginine degrading enzymes mentioned above being injected into the systemic circulation, viz. L-arginasel (rhArg1) and L-arginine deiminase (ADI). Many types of tumour cells cannot convert citrulline into arginine because the two coupled enzymes involved, argininosuccinate synthetase (ASS) and argininosuccinate lyase (ASL), are highly repressed. Both arginine catabolizing enzymes usually give good results with sensitive tumours, which cannot handle citrulline. Although, so far, the lion』s share of the work has been done with ADI, their respective merits have to be carefully considered with regard to their relative efficacy against a broad spectrum of tumours and their side effects. The products of ADI are citrulline (which can readily be converted back to arginine via the unrepressed pathway of ASS/ASL in the kidney), and the toxic by-product nascent ammonia [12]. In contrast, rhArg1 produces ornithine, which is much less quickly converted to arginine via citrulline, and urea (a non-toxic excretory product). We will see below why ADI is, for other reasons, less favourable than rhArg1.
Both of these arginine catabolizing enzymes usually give good results with sensitive tumours, which are those unable to convert citrulline toarginine
One drawback is that no enzyme lasts long when injected into the systemic circulation, usually having a t1/2 of 20-30 min. it would be subject to degradation by proteases, and of foreign, elicits an immune response, eventually being neutralized by antibodies after repeated injections (as found with repeated L-arginase treatment). ADI is a mycoplasma enzyme, and therefore antibodies will inevitably be produced against it; in contrast arginase 1 is a natural enzyme in man found in the liver. But proteases will attack rhArg1 as well as ADI, and to protect the enzymes, they both have to be pegylated by attaching short polymer chains of either 5KDa or 20KDa of polyethylene glycol (PEG) to the proteins through exposed ԑ-amino groups of lysine residues, a process referred to as 「pegylation」. PEG is expensive and currently in short supply worldwide in the form required to effect the best pegylation of rhArg1 (BCT100 is rhArg15,000peg). BCT has developed a PEG facility for the synthesis and linking of the chains covalently to proteins. This work is of high GMC standard and has been specifically designed for the purpose of producing large quantities of PEG that are anticipated to be required in future clinical trials. We also use the less toxic linkage procedure through a succinimidyl group rather than via the cyanuric acid coupling method.
A new dimension: a second urea cyclt enzyme deficiency in cancer cells
In the absence of the ASS/ASL (a coupled pathway) of the urea cycle, cells become auxotrophic gor arginine [5]. The tumour types mentioned above are typically of this kind, but resistance is found or can develop, usually associated with a several hundred-fold increase in ASS and ASL expression. Although initially it was reported that 11/11 hepatocellular carcinoma cell lines were ASS/ASL-negative and highly sensitive to ASI, our data has found no less than 5 HCC cell lines that have normal levels of these urea cycle enzymes. All 5 lines proved to be resistant to ADI [13]. Two ASS/ASL highly positive cells lines (one colorectal carcinoma, the other lung carcinoma) were also tested, and both proved to be resistant to ADI. It was expected on this basis that none of our 5 ASS/ASL positive lines (including Hep3B and HepG2) would succumb to rhA4g1 treatment, and yet all 5 did, although the 2 non-HCC ASS/ASL-positive lines remained resistant. This presented an apparent paradox which was resolved when it was found that these 5 HCC cell lines were deficient in the conversion of ornithine to citrulline andhence arginine, because ornithine transcarbamylase (OTC – see box) was not expressed (rhArgl』s product is ornithine, not citrulline). To confirm this finding, the two sensitive HCC lines to rhArg1 mentioned above were transfected with the OTC gene and the enzyme was then expressed in normal quantities; these cells became resistant to rhArg1 in the same way that they were resistant to ADI. There is little doubt that to have control over two points in the flow of intermediates through the urea cycle, as well as attacking arginine directly, is going to be highly beneficial in the treatment of OTC- and ASS/ASL-defective tumours.
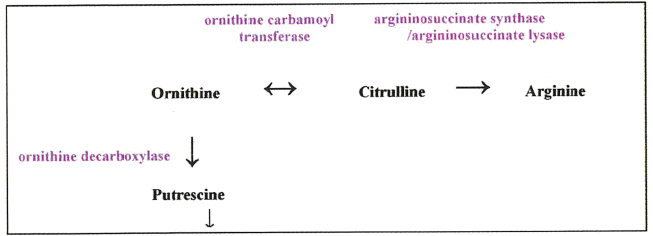
More work is being done on OTC by Professor Andrei Sibirny (Institute of Cell Biology, Lviv, Ukraine). Knowing that OTC is a reversible enzyme, his group is finding methods in vitro and in vivo whereby citrulline can be sent in the opposite cirection through the urea cycle to raise ornithine levels so that the citrulline cannot supply ASS/ASL. It can, however, boost the supply of ornithine as a substrate for another enzyme, ornithine decarboxylase, which produces putrescine, a precursor of the polyamine synthesis pathway.